In a a previous article I mentionned the Protein Data Bank (PDB) that contains the atomic coordinates of some RNAs. The question now is: what kind of information can we extract from a PDB file?
We’re gonna take a close look to two kind of information:
- dihedral angles
- interactions between bases
Dihedral angles
What is a dihedral angle?
In chemistry, a dihedral angle is the angle between planes through two sets of three atoms, having two atoms in common.

Dihedral angle (chemistry)
Dihedral angle \(\phi\) formed by two sets of three atoms: (blue and purple) and (purple and red).
Dihedral angles in RNA
Now that you know what a dihedral angle is, let’s jump to what is a dihedral angle for RNAs! As you can see in the figure below, there is quite a number of dihedral angles.

Dihedral angle (RNAs)
RNA backbone where torsion angles are tagged at the center of the bond forming the dihedral angles. In blue, the 6 dihedral angles of RNA (\(\alpha, \beta, \gamma, \delta, \varepsilon, \zeta\)) and in purple (\(\chi\)) the rotation angle of the base. (Inspired by Stephen Neidle. Principles of Nucleic Acid Structure. Academic Press, Oct. 2007. isbn: 978- 0123695079.)
As you probably notice, the definition of the \(\chi\) is a bit fuzzy: it depends of the base, whether it is a pyrimidine or a purine.
For the sake of clarity, here’s the complete definition of dihedral angles:
alpha : O3'(i-1) - P(i) - O5'(i) - C5'(i)
beta : P(i) - O5'(i) - C5'(i) - C4'(i)
gamma : O5'(i) - C5'(i) - C4'(i) - C3'(i)
delta : C5'(i) - C4'(i) - C3'(i) - O3'(i)
epsilon: C4'(i) - C3'(i) - O3'(i) - P(i+1)
zeta : C3'(i) - O3'(i) - P(i+1) - O5'(i+1)
chi : O4'(i) - C1'(i) - N1(i) - C2(i) -- for pyrimidines
chi : O4'(i) - C1'(i) - N9(i) - C4(i) -- for purines
eta : C4'(i-1) - P(i) - C4'(i) - P(i+1)
theta : P(i) - C4'(i) - P(i+1) - C4'(i+1)
You may have notice that I introduced two more angles: \(\eta\) and \(\theta\). Those were defined by Olson (Configurational statistics of polynucleotide chains. A single virtual bond treatment., Olson, W. K., Macromolecules, 1975) in order to have an idea of the interbase flexibility (and it works!).
Interaction types
When you think about interactions types, two things come in mind:
- stacking interactions
- edge-to-edge interactions
I’m gonna focus on edge-to-edge interactions.
If quite everyone has heard of Watson-Crick/Watson-Crick interactions, they only represent a small part of interaction types. Indeed, in the Léontis and Westhof classification, there exist 12 types of interactions, as shown in the figure below:
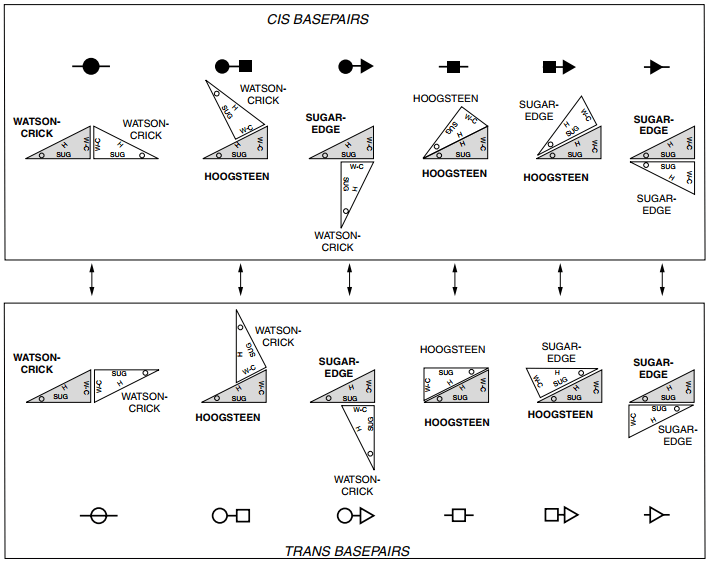
12 types of interactions
12 types of interactions (from Geometric nomenclature and classification of RNA base pairs, Leontis, Neocles B. and Westhof, Eric, RNA, 2001)
Where do those 12 types come from? As you can see in the figure below, interactions between bases are not only on the “Watson-Crick” edges, but can also be on the Hoogsten edge and the sugar edge. This means there are three edges that can interact with one another (as a first approximation).
So, that’s when you tell me that \(\binom{3}{2} = 6 \neq 12\) and I answer that the bond can be cis or trans, as explained in the figure below and so we have \(\binom{3}{2} \binom{2}{1} = 12\).

Base edges
Base edges (from Geometric nomenclature and classification of RNA base pairs, Leontis, Neocles B. and Westhof, Eric, RNA, 2001)
In practice, only 11 of those 12 types of interaction have been found in PDB structures, the missing family being the cis Hoogsten/Hoogsten one.
How do I retrieve the data?
It is possible to extract arc-annotated sequences from PDB files thanks to the software rnaview.
I’m gonna give view some Python code that will allow you to retrieve the dihedral angles and the RNA planarization.
Kept and rejected interactions during planarization
import csv
import re
import sys
import os
sys.path.append("../python/RNA")
import RNAParser
import RNASecondaryStructure
fieldnames = ["PDB.ID", "Full.ID", "Length", "Total", "Structure method",
"Resolution", "K_minus_cis", "K_plus_cis","K_W_W_cis",
"K_W_W_tran", "K_H_H_cis", "K_H_H_tran", "K_S_S_cis",
"K_S_S_tran", "K_W_S_cis", "K_W_S_tran", "K_W_H_cis",
"K_W_H_tran", "K_H_S_cis", "K_H_S_tran", "D_minus_cis",
"D_plus_cis", "D_W_W_cis", "D_W_W_tran", "D_H_H_cis",
"D_H_H_tran", "D_S_S_cis", "D_S_S_tran", "D_W_S_cis",
"D_W_S_tran", "D_W_H_cis", "D_W_H_tran", "D_H_S_cis",
"D_H_S_tran"]
keys = [ [("-/-", "cis")], [("+/+", "cis")],
[("W/W", "cis")], [("W/W", "tran")],
[("H/H", "cis")], [("H/H", "tran")],
[("S/S", "cis")], [("S/S", "tran")],
[("W/S", "cis"), ("S/W", "cis")], [("W/S", "tran"), ("S/W", "tran")],
[("W/H", "cis"), ("H/W", "cis")], [("W/H", "tran"), ("H/W", "tran")],
[("H/S", "cis"), ("S/H", "cis")], [("H/S", "tran"), ("S/H", "tran")],
]
with open("/home/chopopope/PDB-RNA/interactions_data.csv", "wb") as csvfile:
# Instanciate writer and writes headers.
writer = csv.DictWriter(csvfile, fieldnames=fieldnames)
writer.writeheader()
for file in os.listdir("~/PDB-RNA/"):
# For every pdb file
# Re-initate RNA PDB parser
parser = RNAParser.PDBParser(annotation=True)
pdb_filename = os.path.join("~/PDB-RNA/", file)
# Retrieve the PDB ID (i.e. the file name)
PDB_ID = re.split("/", pdb_filename)[-1].strip(".pdb")
# Initiate structure
structure = parser.get_structure(file, pdb_filename)
# Retrieve data from header
METHOD = structure.header["structure_method"]
RESOLUTION = structure.header["resolution"]
for model in structure:
# Each structure may have several models
for chain in model.child_list:
# Each model may have several chains
# Retrieves the full-id
FULL_ID = PDB_ID + "_" + str(model.id) + "_" + chain.id
# Retrieves sequence length (useful for stats)
LENGTH = len(chain.get_sequence())
# Computes the secondary structure
plop = RNASecondaryStructure.RNASecondaryStructure(chain)
# Initialization of the dict that will hold the data to store
# in a
# csv file
dico = {}
# Let's start with the easy fields
dico[fieldnames[0]] = PDB_ID
dico[fieldnames[1]] = FULL_ID
dico[fieldnames[2]] = LENGTH
dico[fieldnames[4]] = METHOD
dico[fieldnames[5]] = RESOLUTION
# Now, let's get to work!
for key, field in zip(keys, fieldnames[6:20]):
# i.e. all the "kept" fields
dico[field] = plop.kept_interactions().get(key[0], 0)
if len(key) > [1]:
# i.e. double interaction
dico[field] += plop.kept_interactions().get(key[1], 0)
# Now, let's get to work!
for key, field in zip(keys, fieldnames[20:]):
# i.e. all the "discarded" fields
dico[field] = plop.discarded_interactions().get(key[0], 0)
if len(key) > [1]:
# i.e. double interaction
dico[field] += plop.discarded_interactions().get(key[1], 0)
dico[fieldnames[3]] = sum([dico[i] for i in fieldnames[6:]])
# Writes the dict to the csv file.
writer.writerow(dico)
csvfile.close()Dihedral angles
# This code loads every provided .pdb files (with RNA) and process each of them
# so that we can retrieve the dihedral angles of every single nucleotide
# chain of each model of each structure and store them in a csv file.
#
# PDB_ID | Full_ID | Length | Res_ID | Structure_method | Resolution | alpha | [...]
import csv
import re
import sys
import os
sys.path.append("../python/RNA")
import RNAParser
import time
fieldnames = ["PDB ID", "Full ID", "Length", "Residue ID", "Structure method",
"Resolution", "alpha", "beta", "chi", "delta", "epsilon",
"eta", "gamma","theta", "zeta"]
with open("~/PDB-RNA/dihedrals_data.csv", "wb") as csvfile:
# Instanciate writer and writes headers.
writer = csv.DictWriter(csvfile, fieldnames=fieldnames, quoting=csv.QUOTE_NONNUMERIC)
writer.writeheader()
for file in os.listdir("~/PDB-RNA/"):
# For every pdb file
# Re-initate RNA PDB parser
parser = RNAParser.PDBParser(annotation=True)
pdb_filename = os.path.join("~/PDB-RNA/", file)
# Retrieve the PDB ID (i.e. the file name)
PDB_ID = re.split("/", pdb_filename)[-1].replace(".pdb", "")
# Initiate structure
structure = parser.get_structure(file, pdb_filename)
# Retrieve data from header
METHOD = structure.header["structure_method"]
RESOLUTION = structure.header["resolution"]
for model in structure:
# Each structure may have several models
for chain in model.child_list:
written = False
# Each model may have several chains
# Retrieves the full-id
FULL_ID = PDB_ID + "_" + str(model.id) + "_" + chain.id
# Retrieves sequence length (useful for stats)
LENGTH = len(chain.get_sequence())
data = chain.get_dihedrals()
for residue in data.keys():
# Retrieve the esidue ID
RES_ID = residue.get_id()[1]
# Initialization of the dict that will hold the data to store in a
# csv file
dico = {}
# Let's start with the easy fields
dico[fieldnames[0]] = PDB_ID
dico[fieldnames[1]] = FULL_ID
dico[fieldnames[2]] = LENGTH
dico[fieldnames[3]] = RES_ID
dico[fieldnames[4]] = METHOD
dico[fieldnames[5]] = RESOLUTION
for field in fieldnames[6:]:
# Let's work on the angles
dico[field] = data[residue][field]
# Writes the dict to the csv file.
writer.writerow(dico)
time.sleep(0.1)
csvfile.close()That’s all for today!